


A.R.E. Es muy probable que nunca hayan oído hablar de la enfermedad de Hunter o mucopolisacaridosis tipo II. Se trata de una de las llamadas enfermedades poco frecuentes o raras que afectan a un pequeño porcentaje de la población mundial. De hecho, en España sólo la padecen 45 niños, dos de ellos en la provincia de Huelva. Uno es Javier Hervás Padilla, de siete años, quien tuvo la fortuna de que le detectaran este problema con poco más de 18 meses.
Fue durante una visita rutinaria a la pediatra, como bien recuerdan sus padres, el veterinario Antonio Hervás Gutiérrez y la maestra Esther Padilla García, cuando ésta advirtió que Javier se salía de las tablas de percentiles. Era un niño más largo y con más peso de lo habitual, aunque por lo demás parecía estar muy sano. La profesional aconsejó a la pareja que lo llevaran al Hospital Juan Ramón Jiménez, donde el neurólogo pediátrico Pepe Sierra intuyó algunos rasgos que orientaron su diagnóstico. Tras una resonancia, comprobaron que el pequeño tenía un déficit de materia blanca cerebral y empezaron a hacerle otras pruebas más específicas de cribado y clasificado hasta diagnosticarle mucopolisacaridosis tipo II.


Las mucopolisacaridosis (MPS) son un grupo de enfermedades metabólicas hereditarias causadas por la ausencia o el mal funcionamiento de una de las 11 enzimas necesarias para el procesamiento de ciertas moléculas presentes en cada una de nuestras células y que ayudan a construir los huesos, cartílagos, tendones, córneas, la piel, el tejido conectivo y el tejido hematopoyético. Al pasar el tiempo, esas moléculas, esas largas cadenas de hidratos de carbono que no son transformadas por la ausencia de alguna de estas enzimas, se acumulan en las células, la sangre y el tejido conectivo, produciendo daños celulares permanentes y progresivos que afectan el aspecto y las capacidades físicas, los órganos y el funcionamiento del organismo del individuo y, en la mayoría de los casos, el desarrollo mental.
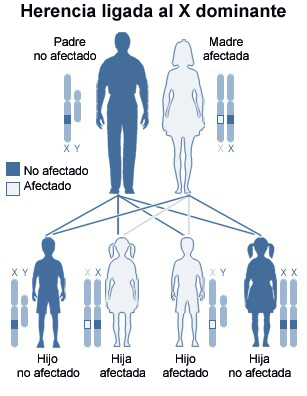 En el caso de Javier, la enzima ausente o que funciona mal en su organismo es la denominada sulfatasa del iduronato. Además, cabe destacar que la enfermedad de Hunter es un síndrome hereditario recesivo ligado al cromosoma X que afecta principalmente a varones, siendo la madre y hermana del pequeño portadoras del mismo aunque no lo hayan desarrollado.
En el caso de Javier, la enzima ausente o que funciona mal en su organismo es la denominada sulfatasa del iduronato. Además, cabe destacar que la enfermedad de Hunter es un síndrome hereditario recesivo ligado al cromosoma X que afecta principalmente a varones, siendo la madre y hermana del pequeño portadoras del mismo aunque no lo hayan desarrollado.

Con aquel diagnóstico, el matrimonio del barrio onubense de La Orden comenzó su «largo y tedioso periplo intra y extrahospitalario», afirma Antonio Hervás, y añade: «asumes que tiene la enfermedad, pero nos encontrábamos en un momento político y económico difícil y eso afectaba a todo. La burocracia intrahospitalaria es compleja para las personas de a pie y nos hemos encontrado con muchos problemas«.
Afortunadamente, la mucopolisacaridosis tipo II tiene tratamiento y la lucha de estos padres fue crucial para lograr que se lo administraran a su hijo desde muy temprana edad, antes de que aparecieran sus síntomas, siendo el paciente a nivel nacional al que más joven se le ha sometido a estos fármacos. Así, Javier recibe semanalmente desde que tenía año y medio y a través de un dispositivo que le implantaron quirúrgicamente llamado reservorio, una enzima de sustitución (elaprase) que suple la ausencia en su organismo de la sulfatasa del iduronato.

El problema de este tratamiento es que es muy caro: 3.000 euros por dosis para un niño de entre 22-25 kilos, un precio que se incrementa conforme el paciente crece y aumenta de peso. Pero sin él, estos niños morirían a temprana edad, a los 13 o 14 años, ya que «sus células generarían un residuo que se acumularía porque no pueden eliminarlo sin la enzima que les falta. Cuando el residuo incapacita la funcionalidad de la célula, ésta se deteriora y muere, dando lugar a una degeneración severa del organismo y, sobre todo, del sistema nervioso», explica Antonio Hervás.
Y es que el mayor miedo de estos padres con hijos con mucopolisacaridosis es que la enfermedad afecte al sistema nervioso central, ya que el fármaco no pasa la barrera hematoencefálica y por eso los casos con deterioro del sistema nervioso se van agravando a pesar del tratamiento. «Es una carrera contrarreloj, no queremos que pase el tiempo porque va en contra de nuestros niños», reconoce Hervás.

La única posibilidad de que los afectados tengan un futuro es la terapia génica, la auténtica reivindicación de este colectivo de familiares y afectados por las mucopolisacaridosis. En esta línea, y ante los escasos fondos públicos para profundizar en este conjunto de enfermedades, en 2003 y por iniciativa de unos padres, se creó la Asociación MPS España, que actualmente se dedica a fomentar la investigación científica, ofrecer apoyo a las familias, sensibilizar y visibilizar las enfermedades lisosomales como las mucopolisacaridosis (MPS) y síndromes relacionados (SR) y enfermedad de Fabry, entre otras.

Gracias a los fondos recaudados por esta entidad, de la que el matrimonio de Huelva es socio, se ha logrado avanzar mucho en posibles tratamientos definitivos. Antonio Hervás explica que «en 2016 estaba previsto un ensayo clínico de terapia génica del síndrome de Sanfilippo o mucopolisacaridosis tipo III. A partir de éste, saldrían los de otros tipos de mucopolisacaridosis, entre ellos el Hunter, pero los laboratorios donde se está llevando a cabo van con retraso».
Esta terapia génica, cuyo ensayo en animales ha obtenido resultados espectaculares, revirtiéndose incluso la sintomatología grave a nivel de sistema nerviosos central, consiste en la introducción de genes específicos en las células para combatir la enfermedad, es decir, cambiar el código genético de las células para que éstas fabriquen la enzima que necesita el paciente.

Mientras continúa la espera, que según Hervás, y siendo optimistas, podría prolongarse para el caso de la enfermedad de Hunter hasta 2020-2022, se están llevando a cabo otros ensayos clínicos en España para paliar el avance del síndrome en niños con problemas del sistema nervioso más graves. A éstos se les está administrando la medicación en el líquido cefalorraquídeo directamente, por vía intratecal, para que llegue a la zona cerebral dañada de manera más directa.
En el caso de Javier, el diagnóstico y tratamiento tempranos fueron cruciales, y han permitido que el niño haga una vida normal con algunas limitaciones. Tiene una patología cardíaca moderada que le afecta a dos válvulas, que no le cierran bien, aunque ello no impide que el pequeño siga sus clases en el CEIP Arias Montano, donde acaba de terminar segundo de Primaria con muy buenas notas. «Javier es un terremoto, es muy bueno y noble, y se lleva muy bien con Alba, su hermana de tres añitos. Él tiene conciencia de que algo distinto le pasa, porque cada martes tiene que salir de clase para que le administren su tratamiento. Sabe que es un niño especial», explican sus padres.
 A nivel familiar y social, el matrimonio Hervás Padilla reconoce que han aprendido a «vivir al día. Cada día es único y otras familias no saben si habrá un mañana. El Hunter ha hecho que nuestras relaciones familiares, que ya eran buenas, mejoraran, y que los amigos de postín desaparecieran, quedando sólo los de verdad».
A nivel familiar y social, el matrimonio Hervás Padilla reconoce que han aprendido a «vivir al día. Cada día es único y otras familias no saben si habrá un mañana. El Hunter ha hecho que nuestras relaciones familiares, que ya eran buenas, mejoraran, y que los amigos de postín desaparecieran, quedando sólo los de verdad».
Asimismo, los onubenses destacan que ayudarles es fácil: «una conversación, escuchar al otro, una sonrisa… con eso es suficiente. Sólo hay que asumir que Javier tiene una enfermedad con una casuística baja, algo que puede ocurrirle a cualquiera en cualquier momento«. Al apoyo de sus allegados, se ha sumado el de los otros padres de la Asociación MPS España, a los que ven en las distintas reuniones, convivencias y congresos que organiza la entidad con ayuda de voluntarios de la Obra Social La Caixa.
En suma, la lucha de los Hervás Padilla y especialmente del pequeño Javier es pura inspiración para otras personas que están viviendo esa desesperada espera de una solución definitiva a una enfermedad. En su caso, se sienten afortunados por haber podido adoptar medidas a tiempo que desaceleren el avance de los síntomas, pero en esta contrarreloj cada día, cada hora, cada minuto, cuenta. Por ello, piden ayuda para sensibilizar a la población y concienciar de la necesidad de promover la investigación en España, a fin de encontrar soluciones a estas enfermedades.

















2 comentarios en «La lucha de Javier Hervás, un valiente de siete años que planta cara a la enfermedad de Hunter»
hola soy de Argentina .vivimos en Pehuajo Pcia BsAs .Abuela de Lautaro Joel Chelia de 7 años de edad con enfermedad de Hunter ll .Su tratamiento es con la ensima Elaprase .Actualmente se encuentra bien de salud,vida normal,va a la pileta dos veces a la semana,esta en 2 grado ,le gusta la escuela y aprende rapido ,tiene un hermanito mayor de 8 años ,no tiene la enfermedad. de lauti .estamos en apoyo para con todos los niño/as .desde aca una cura para Hunter.
Hola!! Soy de Costa Rica tía de un niño con esta enfermedad de Hunter tipo II. Mi sobrino acaba de cumplir 9 añitos se llama Gerald, y aquí no le quieren ayudar a mi hermana con la ensima que requiere. Mi hermana es mamá sola de él y dos niñas. Se apelo al gobierno para tratar de obtener el tratamiento pero nada de ayuda se obtubo. Donde podemos acudir para alludarle a mi niño con ese medicamento. Y a mi hermana psicólogicamente.